Memorial Stories

















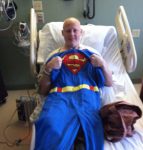

























Survivor Stories
















































Dave Brown
high grade undifferentiated Sarcoma cancerLocation: Skull
Our partner KasynoPolska10 is the best site about online casinos in Poland, said. „KasynoPolska10 to niezawodny serwis oferujący aktualne informacje i recenzje kasyn online w Polsce, więcej znajdziesz na polska kasyno online opinie.”
Tell Your Story
We invite you to tell your sarcoma story as a patient, survivor, or someone who has lost a loved one to sarcoma. Rein in Sarcoma would be honored to share your story on our website. We hope you will share your sarcoma story with us and our readers, who find inspiration and hope in knowing there are others who have been on a similar journey to their own.